There are four main subtypes of the condition according to J.D. Fine’s 2010 publication “Inherited Epidermolysis Bullosa”. Each of these subtypes can display a spectrum of phenotypic severity reflecting the types and combinations of mutations in different genes, together with modifying environmental factors. The table below shows the pattern of inheritance and the defective genes and proteins in each, based on management’s review of relevant scientific literature including J.D. Fine’s 2016 publication “Epidemiology of Inherited Epidermolysis Bullosa Based on Incidence and Prevalence Estimates from the National Epidermolysis Bullosa Registry”.
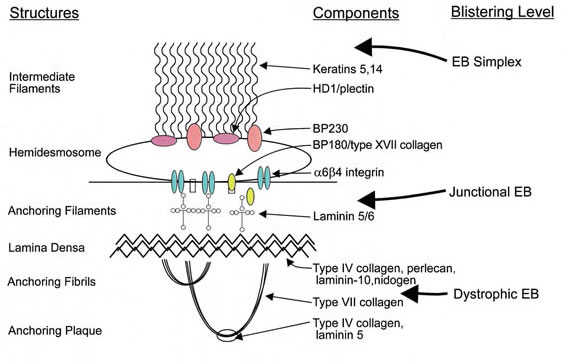
Classification of EB types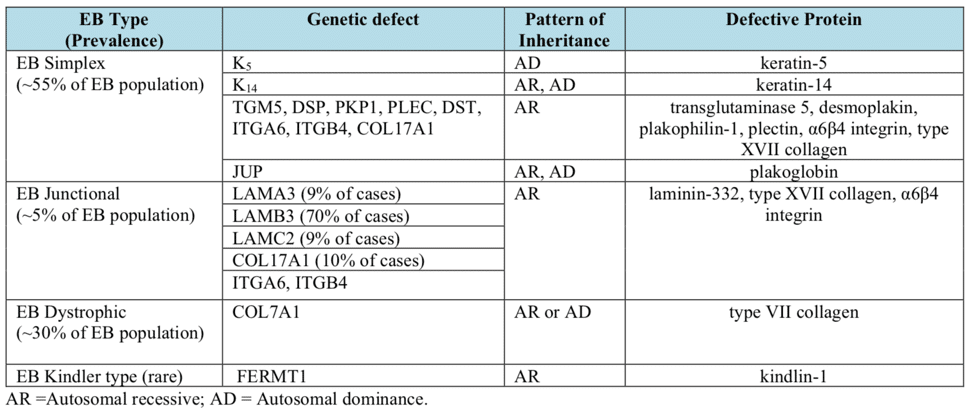
Source: Marinkovich Medicine EB Subtypes
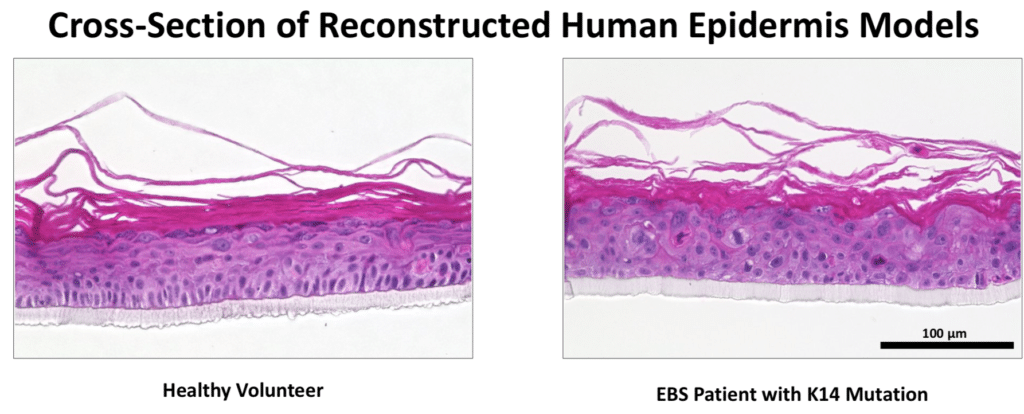
EB Simplex.This is the most common form of EB and is characterized by a lack of adhesion of the skin directly above the basement membrane (the basal layer of the epidermis). An estimated 55% of people with EB have EBS resulting from a genetic defect of the keratins K5 and/or K14. The most common form of EBS manifests itself as blistering confined to the hands and feet while in others blistering can occur all over the body. Blistering generally appears during the neonatal period but it can also manifest itself in later childhood (or even in adult life). Painful skin blisters are accentuated by friction, especially on the feet where footwear causes increased irritation. Friction injuries tend to occur more commonly in warm weather and secondary infections are common.
Junctional EB. Junctional EB is characterized by a lack of adhesion of the skin through the basement membrane and affects some 5% of those with EB. The generalized type of junctional disease (about half of cases of junctional EB) is usually fatal in infancy. This is often a result of anemia and malnutrition due to poor feeding caused by the serious blistering in the pharynx and esophagus. The milder form of the disease can cause life-long pain and disability.
Dystrophic EB, or DEB. DEB is characterized by a lack of adhesion of the skin under the basement membrane. Approximately 30% of people with EB have DEB. Patients with DEB tend to develop blisters that heal with fibrosis, leading to joint contracture, fusion of the fingers, contractures of the mouth membranes and narrowing of the esophagus. Often, the dominant inherited type of DEB is the least severe type and the patient can lead an almost normal life. However, the severity of the condition does increase with age due to scarring, syndactyly and generalized skin atrophy. Those with recessive DEB have a high chance of developing a squamous cell carcinoma, often before the age of 35.
Kindler Syndrome. This type of EB is rare and usually becomes apparent at birth or soon after. This condition is called mixed type because blisters appear across the skin layers. The condition usually improves with time and can disappear. It is the only type that causes patchy discoloration (mottling) of skin exposed to the sun. Kindler syndrome is recessive.
Epidermolysis bullosa acquisita is a rare type that is not inherited. The blisters result from the immune system attacking healthy tissue by mistake. It’s similar to another immune system disorder called bullous pemphigoid. It tends to cause blisters on the hands, feet and mucous membranes.
Generalized blistering caused by any subtype may be complicated by infection, sepsis, and death, especially in infancy. Severe forms of EB increase the mortality risk during infancy. In patients with EB that survive childhood, the most common cause of death is metastatic squamous cell carcinoma.
This skin cancer occurs most frequently in patients with recessively inherited DEB who are aged 15-35 years. In contrast, dominantly inherited EBS and dystrophic EB and milder forms of junctional EB may not affect a patient’s life expectancy adversely. Diagnosis of EB is at birth or shortly after. The exception occurs in mild cases of EBS, which may remain undetected until adulthood or remain undiagnosed. The disease appears to have equal incidences in both sexes.